Deficiencia de alfa-1 antitripsina
Sinónimos en un sentido más amplio
- Síndrome de Laurell-Eriksson
- Deficiencia del inhibidor de la proteasa alfa-1
Español: deficiencia de alfa1-antitripsina
Introducción
La deficiencia de alfa-1-antitripsina significa, como su nombre indica, la falta de la proteína alfa-1-antitripsina, que se forma en los pulmones y el hígado. Entonces es un trastorno metabólico.
Esta condición se hereda de forma autosómica recesiva. Ocurre con una frecuencia de 1: 1000 a 1: 2500 en la población.

causas
La causa de la deficiencia de alfa-1 antitripsina radica en una falla en la herencia.
La deficiencia de la proteína alfa-1-antitripsina se hereda de forma autosómica recesiva. Esto significa que la enfermedad se hereda independientemente del género y solo se manifiesta realmente si hay dos copias genéticas defectuosas. Ambos padres deben estar enfermos o actuar como portadores de la información genética. Solo un gen que lleva la información incorrecta no puede causar ningún daño. El defecto está en el cromosoma 14. En este cromosoma está el gen que se utiliza en individuos sanos para la síntesis (Fabricación) de alfa-1 antitripsina es responsable.
La alfa-1-antitripsina es una proteína producida por el cuerpo (proteína), que se produce principalmente en las células del hígado.
Tiene la función de inhibir las enzimas que dividen las proteínas. Una deficiencia de alfa-1-antitripsina ahora conduce a una actividad excesiva de estas enzimas que dividen las proteínas. Esto da como resultado la descomposición del propio tejido del cuerpo. Su tarea más importante es inhibir la enzima elastasa leucocitaria. Esto a saber, rompe la elastasa en la pared de los alvéolos.
Síntomas y dolencias.
Dado que la producción de alfa-1-antitripsina tiene lugar principalmente en los pulmones y el hígado, aquí también se producen daños y deficiencias. Allí también se produce la degradación del propio tejido del cuerpo.
Existe una variabilidad muy amplia en la forma. En personas con daño pulmonar severo, la afectación hepática es sorprendentemente rara y viceversa. La distribución por edades también es bastante diferente. Si bien algunos tienen enfermedades pulmonares en etapa terminal en la tercera a la quinta década de la vida, otros no tienen ningún daño en los pulmones a la edad de 30 años.
Síntomas en la piel
A veces, los pacientes con deficiencia de alfa-1 antitripsina tienen inflamación en la grasa subcutánea. Este es demarcado y rojizo. Se llama paniculitis. También hay otras causas de esta inflamación. El mecanismo exacto de su formación aún no se conoce. Esta inflamación localizada puede ser muy persistente y dolorosa.
Otro síntoma en la piel es el color azul (cianosis). Esto se debe a una saturación insuficiente de oxígeno de la sangre cuando los pulmones están afectados, como el enfisema. No solo la piel tiene un tinte azulado, sino también las membranas mucosas y la lengua. La cianosis se presenta en muchos cuadros clínicos, por lo que no es específica de la deficiencia de alfa-1 antitripsina.
Síntomas y consecuencias en los pulmones.
La proteína alfa-1-antitripsina no solo se encuentra en el hígado, sino también en los pulmones. Aquí también juega un papel importante en la buena función pulmonar. Si esta alfa-1-antitripsina es deficiente, los componentes importantes de los pulmones se degradan, lo que resulta en la destrucción constante del tejido pulmonar.
Una deficiencia de alfa-1 antitripsina causa enfisema en los pulmones. El enfisema pulmonar es una inflación excesiva de los pulmones. Esto ocurre debido a los cambios inflamatorios en la estructura pulmonar. Las paredes de los alvéolos ya no son lo suficientemente estables y se destruyen por degradación enzimática. Esto crea grandes cavidades en los pulmones de las que ya no puede escapar el aire inhalado. Por eso se habla de sobreinsuflación de los pulmones.
La enfermedad pulmonar obstructiva crónica (EPOC) también se desarrolla en la edad adulta temprana. El intercambio de gases en los pulmones se altera, lo que provoca una falta de oxígeno en la sangre. La tos con esputo es típica de la EPOC.
La sensación de falta de aire también es típica en la etapa avanzada. Esto también puede tener consecuencias para el corazón, por lo que también se daña. En el caso de daño pulmonar muy avanzado y fracaso de otras medidas terapéuticas, un trasplante de pulmón puede ser una medida necesaria.
Leer más sobre: EPOC en etapa terminal
Síntomas y consecuencias en el hígado.
El hígado es el primer órgano afectado por la deficiencia de alfa-1 antitripsina. La proteína alfa-1-antitripsina está alterada. La forma de la proteína es diferente de la forma sana. Esto lleva al hecho de que se acumula en las células del hígado y no se puede liberar correctamente. Esto crea un defecto.
En los recién nacidos homocigotos (es decir, que tienen dos copias genéticas defectuosas) de la enfermedad, el hígado ya está dañado en la infancia. Se les diagnostica ictericia neonatal prolongada (ictericia = coloración amarillenta de la piel y la esclerótica (blanco de los ojos)).
Si la enfermedad solo aparece en la edad adulta (aprox. 10-20%), se acompaña de hepatitis crónica (inflamación del hígado) y cirrosis hepática posterior.
Además, aumenta el riesgo de desarrollar cáncer de hígado (carcinoma hepatocelular). La cirrosis del hígado puede provocar muchas complicaciones para los afectados. En una etapa avanzada, la esperanza de vida también se reduce significativamente.
Leer más sobre el tema: Cirrosis del higado
diagnóstico
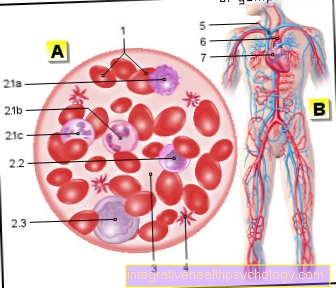
El diagnóstico de deficiencia de alfa-1 antitripsina se basa en una muestra de sangre y un examen de laboratorio. La sangre del paciente se examina para determinar sus componentes individuales (en este caso, especialmente para la composición de proteínas).
Hay una ausencia casi completa de proteínas alfa-1. Las enzimas hepáticas elevadas también se pueden encontrar en la sangre. La ecografía muestra un hígado agrandado (med.: Hepatomegalia).
Una biopsia de hígado (muestras de tejido del hígado) también muestra depósitos característicos.
¿Cómo son los valores hepáticos con una deficiencia de alfa-1-antitripsina?
Dado que la enzima alfa-1-antitripsina no se forma correctamente en el hígado, la enzima formada incorrectamente se deposita en las células del hígado y, por lo tanto, las destruye.
Esto aumenta los marcadores del parénquima hepático como GOT, GPT y glutamato deshidrogenasa (GLDH). La fosfatasa alcalina también suele estar aumentada. Con la cirrosis hepática avanzada, también se ven afectados otros parámetros. Lo típico sería una albúmina disminuida, una esterasa de colesterol disminuida (CHE) y factores de coagulación disminuidos, así como un valor de amoníaco aumentado.
Leer más sobre esto: Valores hepáticos elevados
¿Qué prueba puede detectar la deficiencia de alfa-1 antitripsina?
Hay dos pruebas que realmente prueban esta condición. Esa es la electroforesis en suero y la prueba genética.
En la electroforesis sérica, se determina la concentración total de proteínas séricas de la sangre y el fraccionamiento de estas. Es una prueba de diagnóstico de laboratorio. En general, las concentraciones de proteínas se muestran como una línea con picos en un sistema de coordenadas. Hay 5 picos, el segundo pico de esta curva muestra el contenido de globulinas alfa-1, que incluye alfa-1 antitripsina. Si hay una deficiencia, este pico es correspondientemente menor.
La prueba genética se lleva a cabo, por ejemplo, en un laboratorio genético humano. Para hacer esto, se examina el ADN del paciente en busca de mutaciones en el gen asociado (ver herencia).
Todas las demás pruebas, como una prueba de función pulmonar, una radiografía de tórax o una ecografía del hígado, pueden explicar los síntomas de la enfermedad, pero no la causa.
Este artículo también puede interesarle: La prueba de alfa-1 antitripsina.
terapia
La deficiencia de alfa-1-antitripsina ahora se puede remediar fácilmente administrando la proteína por vía intravenosa.
Además, sin embargo, se deben tratar las enfermedades de los órganos (especialmente la cirrosis hepática) y se debe reparar cualquier daño que ya haya ocurrido. En casos extremos, sin embargo, se debe considerar un trasplante de hígado o pulmón.
La administración de alfa-1-antitripsina tiene los siguientes efectos secundarios:
- náusea
- Alergias
- fiebre
- Raras: shock anafiláctico (shock alérgico), que puede poner en peligro la vida
La terapia genética está en perspectiva en el futuro.
Esperanza de vida
La deficiencia de alfa-1 antitripsina se debe a varias mutaciones en los genes. Es una enfermedad hereditaria poco común que se presenta con una frecuencia de aproximadamente 1: 2000 a 1: 5000.
Los afectados pueden sufrir una forma leve o grave de la enfermedad, que se asocia con diversas enfermedades y complicaciones secundarias.La esperanza de vida, especialmente de aquellos pacientes que se ven afectados por una forma grave, se reduce en comparación con la población sana. La esperanza de vida se estima en unos 60 a 68 años.
Sin embargo, esto solo se aplica a los afectados que realizan una terapia constante y se adhieren a una estricta prohibición de fumar. También se debe evitar el consumo de alcohol, ya que aumenta la probabilidad de que ocurra una enfermedad hepática.
La esperanza de vida depende en gran medida de las enfermedades secundarias y de la función orgánica conservada de los pulmones y el hígado. En caso de insuficiencia orgánica o función severamente restringida, el último recurso suele ser un trasplante de órgano, que también se asocia con una esperanza de vida reducida y el riesgo de complicaciones adicionales.
profilaxis
No existe una profilaxis real porque la enfermedad se hereda. Los afectados no deben fumar, ya que lo hace más difícil y ejerce aún más presión sobre los pulmones. El alcohol también debe evitarse debido a la tensión en el hígado.
¿La deficiencia de alfa-1 antitripsina es hereditaria?
La deficiencia de alfa-1 antitripsina es hereditaria. La secuencia de genes correspondiente de esta enzima está en el cromosoma 14.
Si la secuencia del gen contiene una mutación, la secuencia ya no se puede leer correctamente y la enzima se forma incorrectamente. Por tanto, la gravedad de la enfermedad es variable. La mutación se hereda, es decir, se transmite de la madre o el padre. Un paciente está completamente desarrollado cuando un defecto se hereda tanto del lado paterno como del materno. El grado de afectación del paciente depende de la genética, pero también de factores externos como el tabaquismo.
Anatomía y ubicación de los pulmones.
- Pulmón derecho
- TráqueaTráquea)
- Bifurcación traqueal (carina)
- Pulmon izquierdo
Resumen
La deficiencia de alfa-1 antitripsina es una enfermedad metabólica hereditaria que produce principalmente cambios en el tejido pulmonar. La frecuencia de la enfermedad es 1: 2000. Debido a la falta de esta enzima, no existe un efecto inhibidor sobre las enzimas que dividen las proteínas.
Debido a esta deficiencia, su propio tejido pulmonar se descompone o se digiere.
Se produce enfisema pulmonar (que incluye tos y dificultad para respirar) y, con afectación adicional del hígado (10-20%), hepatitis (ictericia). El diagnóstico se realiza mediante un análisis de sangre. La terapia es mediante terapia de sustitución, es decir, la alfa-1-antitripsina se administra artificialmente. La proteína faltante se administra por vía intravenosa (a través de la vena). No hay profilaxis.