Sarcoma de Ewing
Toda la información proporcionada aquí es solo de naturaleza general, ¡la terapia tumoral siempre está en manos de un oncólogo experimentado!
Sinónimos
Sarcoma de hueso, PNET (tumor neuroectodérmico primitivo), tumor de Askin, sarcoma de Ewing - hueso
Inglés: sarcoma de Ewing
definición
En el cual Sarcoma de Ewing es una de Médula ósea saliente Tumor óseoque puede ocurrir entre las edades de 10 y 30. Sin embargo, afecta principalmente a niños y jóvenes hasta los 15 años. El sarcoma de Ewing es menos común que eso Osteosarcoma.
El sarcoma de Ewing se localiza en los largos Huesos largos (Fémur (Hueso del muslo) y Tibia (Shin)), así como en la pelvis o las costillas. En principio, sin embargo, todos los huesos del tronco y el esqueleto de las extremidades pueden verse afectados, las metástasis especialmente en el Livianos es posible.
frecuencia
La probabilidad de desarrollar sarcomas de Ewing es <1: 1.000.000. Los estudios han demostrado que por cada millón de personas que viven allí, alrededor de 0,6 nuevos pacientes desarrollan sarcomas de Ewing cada año.
En comparación con el osteosarcoma (aproximadamente el 11%) y el condrosarcoma (aproximadamente el 6%), el sarcoma de Ewing ocupa el tercer lugar como otro representante de los tumores óseos malignos primarios. Si bien el sarcoma de Ewing se presenta principalmente entre los 10 y los 30 años, una manifestación principal podría establecerse en la segunda década de la vida (15 años). Por lo tanto, se manifiesta principalmente en el esqueleto en crecimiento, y los niños (56%) padecen sarcoma de Ewing un poco más a menudo que las niñas. Si se comparan los tumores óseos malignos primarios de niños y adolescentes, el sarcoma de Ewing ocupa el segundo lugar: en los sarcomas óseos infantiles, la proporción de los llamados osteosarcomas es de alrededor del 60%, la proporción de sarcomas de Ewing alrededor del 25%. .
causas
Como ya se ha explicado y presentado en el contexto del resumen, la causa que es responsable del desarrollo de la Sarcoma de Ewing no se puede aclarar completamente. Sin embargo, se encontró que los sarcomas de Ewing a menudo ocurren cuando hay anomalías esqueléticas familiares o pacientes menores de uno desde el nacimiento. Retinoblastoma (= tumor de retina maligno que aparece en la adolescencia). La investigación ha demostrado que las células tumorales de la llamada familia de sarcomas de Ewing tienen un cambio Cromosoma # 22 exposición. Se cree que esta mutación está presente en alrededor del 95% de todos los pacientes.
localización
Las ubicaciones más comunes del sarcoma de Ewing se pueden encontrar en los huesos tubulares largos, especialmente en la tibia y el peroné, o en huesos planos. Sin embargo, como cáncer de hueso maligno, el sarcoma de Ewing puede afectar a todos los huesos. Los huesos más grandes son los más afectados, los más pequeños rara vez. Si los huesos tubulares largos se ven afectados, el tumor generalmente se encuentra en el área de la llamada diáfisis, el área del eje.
Lugares preferidos:
- aprox.30% fémur (hueso del muslo)
- aprox.12% tibia (espinilla)
- aprox.10% húmero (hueso de la parte superior del brazo)
- aprox.9% lavabo
- aprox.8% peroné (peroné).
Debido a la metástasis hematógena grave que se produce al principio (ver la siguiente sección), también es concebible la localización en los tejidos blandos.
Localización en la pelvis
El sarcoma de Ewing se localiza en el hueso pélvico como el tumor primario (sitio de origen del tumor) solo en aproximadamente uno de cada cinco casos. Sin embargo, con mucha más frecuencia, el tumor primario se localiza en un hueso tubular largo.
Los primeros síntomas pueden ser hinchazón, dolor y sobrecalentamiento en la zona pélvica.
Localización en el pie
El pie es una ubicación poco común en un tumor primario. Es más común que los tumores primarios de la tibia o el peroné favorezcan una metástasis en el pie.
Si hay una hinchazón dolorosa poco clara y un sobrecalentamiento del pie, especialmente en la adolescencia, debe excluirse el sarcoma de Ewing además de la artritis juvenil. Aquí no es necesario asumir lo peor. Los diagnósticos específicos en forma de imágenes pueden proporcionar una claridad inicial sobre las causas de las quejas.
metástasis
Como se mencionó anteriormente, esto se aplica sarcoma de Ewing como metástasis hematógena temprana (= a través del torrente sanguíneo). Por tanto, las metástasis también pueden asentarse en los tejidos blandos. De estos, principalmente es el pulmón afectado. Sin embargo, el esqueleto también puede verse afectado por metástasis a través del torrente sanguíneo.
El hecho de que el sarcoma de Ewing se clasifique como metastásico temprano lo demuestran los estudios que muestran que las metástasis pueden detectarse en alrededor del 25% de todos los casos en el momento del diagnóstico. Dado que, lamentablemente, las metástasis no siempre se pueden descubrir, es probable que la tasa de oscuridad sea mucho mayor.
diagnóstico
Los sarcomas de Ewing pueden causar una variedad de síntomas. Deben enumerarse a continuación:
- Dolor de causa desconocida
- Hinchazón y generalmente dolor en la (s) región (es) afectada (s)
- Hinchazón de los ganglios linfáticos
- signos locales de inflamación (enrojecimiento, hinchazón, sobrecalentamiento)
- pérdida de peso no deseada
- Restricciones funcionales hasta la parálisis
- Fractura sin accidente
- Sudores nocturnos
- leucocitosis moderada (= aumento del número de leucocitos en la sangre)
- rendimiento disminuido
Se puede descartar un tumor con suficiente probabilidad si se cumplen los siguientes criterios de acuerdo con los diagnósticos clínicos, de imagen y de laboratorio:
- No hay evidencia de masa
o
La hinchazón visible, la masa comprobada o las quejas poco claras pueden explicarse y documentarse claramente por una enfermedad no tumoral.
Diagnóstico básico:
En principio, los métodos de imágenes se utilizan para diagnósticos básicos. estos son
Examen de rayos x
Examen de rayos X en el área de localización del tumor (al menos 2 niveles)
Ecografía
Ecografía del tumor (especialmente si se sospecha un tumor de tejidos blandos en el diagnóstico diferencial)
Para obtener información adicional y permitir delimitaciones de diagnóstico diferencial, se utilizan diagnósticos de laboratorio (examen de valores de laboratorio). Los siguientes valores se determinan como parte de este diagnóstico de laboratorio:
- Conteo de glóbulos
- Hierro (porque disminuyó en los tumores)
- Electrolitos (para descartar hipercalcemia)
- ESR (velocidad de sedimentación)
- CRP (proteína C reactiva)
- fosfatasa alcalina (aP)
- específico de hueso (AP)
- Fosfatasa ácida (sP)
- Antígeno prostático específico (PSA)
- Ácido úrico (HRS): aumentado con un alto recambio celular, p. Ej. en hemoblastosis
- Proteína total: disminuida en los procesos de consumo
Electroforesis de proteínas - Estado de la orina: paraproteínas - evidencia de mieloma (plasmocitoma)
- Marcador tumoral NSE = enolasa neuronal específica en el sarcoma de Ewing
Diagnóstico especial de tumores
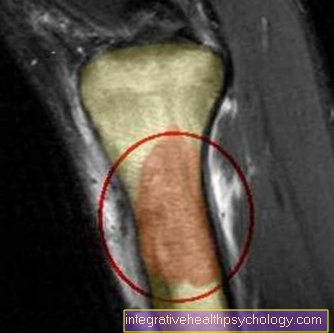
Imágenes por resonancia magnética (IRM)
Además de los métodos de formación de imágenes mencionados en el contexto del diagnóstico básico, la tomografía por resonancia magnética representa una posibilidad adicional que se puede utilizar en casos individuales.
Utilizando MRI (tomografía por resonancia magnética), el tejido blando se puede mostrar particularmente bien, por lo que se puede mostrar la expansión del tumor a las estructuras vecinas (nervios, vasos) de los huesos afectados. Además, el volumen del tumor se puede estimar usando MRI (tomografía por resonancia magnética) y se puede aclarar la extensión local del tumor.
Tan pronto como se sospeche un tumor óseo maligno, se deben tomar imágenes de todo el hueso portador del tumor para descartar metástasis (asentamientos malignos).
Tomografía computarizada (TC):
(especialmente para mostrar estructuras óseas duras (corticales))
Tomografía por emisión de positrones (PET)
(Valor aún no suficientemente válido)
Leer más sobre el tema: Tomografía de emisión de positrones
Angiografía por sustracción digital (DSA) o angiografía para visualizar los vasos del tumor
Gammagrafía esquelética (gammagrafía trifásica)
biopsia
Como se mencionó varias veces anteriormente, la distinción entre sarcoma de Ewing y osteomielitis, por ejemplo, puede ser bastante difícil. Además del hecho de que los síntomas son similares, la radiografía como tal no siempre puede proporcionar información directa. Si, después de los llamados diagnósticos no invasivos descritos anteriormente, todavía existe la sospecha de un tumor o si el tipo y la dignidad de un tumor no están claros, se debe realizar un examen histopatológico (= examen de tejido).
Procedimientos abiertos
Biopsia por incisión
Como parte de la llamada biopsia incisional, el tumor se expone parcialmente quirúrgicamente. Finalmente, se toma una muestra de tejido (si es posible hueso y tejido blando). El tejido tumoral extraído se puede evaluar directamente.
Biopsia por escisión (extirpación completa del tumor)
Solo se considera en casos excepcionales, por ejemplo, si existe sospecha de malignidad (cambio de tumor benigno a maligno) de osteocondromas más pequeños.
terapia
El enfoque terapéutico aquí suele ser de varios niveles. Por un lado, el llamado plan de terapia suele prever un tratamiento quimioterápico preoperatorio (= quimioterapia neoadyuvante). Incluso después de la extirpación quirúrgica del sarcoma de Ewing, se proporciona tratamiento de seguimiento terapéutico mediante radioterapia y, si es necesario, quimioterapia renovada. Aquí se nota una diferencia con el osteosarcoma: en comparación con el sarcoma de Ewing, el osteosarcoma tiene una menor sensibilidad a la radiación.
Objetivos de la terapia:
Un enfoque terapéutico llamado curativo (curativo) se administra especialmente en pacientes cuyo sarcoma de Ewing está localizado y no muestra metástasis. Mientras tanto, la denominada quimioterapia neoadyuvante en combinación con cirugía y radioterapia abre nuevas oportunidades. Si el sarcoma de Ewing hace metástasis fuera de los pulmones (= enfermedad tumoral generalizada; metástasis extrapulmonares), la terapia suele tener un carácter paliativo (prolongar la vida) (ver más adelante).
Modalidades de terapia:
local:
- quimioterapia preoperatoria
- terapia quirúrgica (resección amplia o radical después de Enneking)
- radioterapia
Sistémico:
quimioterapia antineoplásica
- Terapia de combinación (principalmente (= "primera línea"): doxorrubicina, ifosfamida, Metotrexato / Leucovorina, cisplatino; en la segunda línea (= "segunda línea"): etopósido y carboplatino)
(Los protocolos pueden cambiar con poca antelación)
Terapia curativa:
- quimioterapia agresiva con múltiples sustancias antes y después de la operación
- Tratamiento local en forma de resección quirúrgica del tumor o radiación sola
- Complementar la terapia con Pre-irradiación (por ejemplo, en el caso de tumores inoperables, no respondedores) o mediante post-irradiación
- Es importante mencionar en el contexto de la terapia quirúrgica que, sobre todo debido al mayor desarrollo de los métodos quirúrgicos, las intervenciones que preservan las extremidades son posibles en muchos casos. Sin embargo, la perspectiva de una cura siempre tiene la máxima prioridad, por lo que el enfoque siempre debe estar en la radicalidad (= calidad oncológica) y no en una posible pérdida de función.
- Luego se puede continuar la quimioterapia (ver arriba). Entonces se habla de una supuesta consolidación.
- En pacientes con metástasis pulmonares, pueden ser necesarias intervenciones adicionales en el área de los pulmones, como la extirpación parcial de los pulmones.
Terapia paliativa (que prolonga la vida):
Los pacientes que tienen una enfermedad tumoral generalizada (= metástasis extrapulmonar) deben localizar el tumor primario en el tronco y / o el tumor primario resulta inoperable. En tales casos, generalmente solo es posible la terapia paliativa. En tales casos, la atención se centra generalmente en mantener la calidad de vida, de modo que la terapia se concentra en el alivio del dolor y el mantenimiento de la función.
pronóstico
El hecho de que ocurran o no recurrencias depende en gran medida de la extensión de la metástasis, la respuesta a la quimioterapia preoperatoria y la "naturaleza radical" de la extirpación del tumor. Actualmente se cree que la tasa de supervivencia a cinco años es de alrededor del 50%. Las mejoras operativas en particular han permitido mejorar la probabilidad de supervivencia durante los últimos 25 años.
La tasa de supervivencia disminuye con metástasis primarias. Aquí la tasa de supervivencia ronda el 35%.
Posibilidades de recuperación
Al igual que con otros cánceres, las posibilidades de recuperación del sarcoma de Ewing deben considerarse inicialmente como diferentes individualmente, porque las estadísticas solo muestran las tasas promedio de recuperación y supervivencia.
Las posibilidades de recuperación aumentan si el tumor se puede extirpar quirúrgicamente por completo. Antes de esto, se debe realizar quimioterapia para reducir el tamaño del tumor. Una vez que se ha extirpado quirúrgicamente el tumor, se debe realizar más quimioterapia para destruir cualquier célula tumoral restante.
Si el tumor no se puede extirpar quirúrgicamente por completo, las posibilidades de recuperación son mucho peores. Aquí también debe realizarse un tratamiento de seguimiento con quimioterapia.
Un tumor que no puede operarse definitivamente debe irradiarse.
En general, se puede decir que las perspectivas de curación del sarcoma de Ewing son más precarias si las metástasis ya están presentes en el momento del diagnóstico. Esto significa que el tumor se ha diseminado y también está creciendo en otras partes del cuerpo.
Tasa de supervivencia
Las tasas de supervivencia en general se dan en medicina como el valor estadístico de la "tasa de supervivencia a 5 años". Esto indica en porcentaje qué tan grande es el número de sobrevivientes después de 5 años en un grupo de pacientes definido. La tasa de supervivencia informada para el sarcoma de Ewing se encuentra en un rango entre el 40% y el 60-70%. Estas amplias áreas resultan del hecho de que la tasa de supervivencia depende de la infestación de la región ósea respectiva. Por ejemplo, si los huesos de los brazos y / o las piernas se ven afectados, la tasa de supervivencia a 5 años es del 60-70%. Si los huesos pélvicos están afectados, es del 40%.
¿Qué tan alto es el riesgo de recaída?
La tasa de supervivencia a 5 años promedia el 50%. Aquí se puede suponer que se trata de un cáncer agresivo y maligno. La tasa de supervivencia a 5 años dice que, en promedio, la mitad de todos los sarcomas de Ewing diagnosticados conducen a la muerte.
Sin embargo, si no se pueden detectar más hallazgos después de 5 años después del tratamiento exitoso del sarcoma de Ewing, se dice que el cáncer está curado.
Cura postoperatoria
Recomendaciones:
- en el año 1 y 2:
Se debe realizar un examen clínico cada tres meses. Como regla general, una radiografía local, pruebas de laboratorio, una tomografía computarizada del tórax y se realizó una gammagrafía esquelética de cuerpo completo. Por lo general, se realiza una resonancia magnética local una vez cada seis meses. - en el año 3 al 5:
Se debe realizar un examen clínico cada seis meses. Como regla general, una radiografía local, pruebas de laboratorio, una tomografía computarizada del tórax y se realizó una gammagrafía esquelética de cuerpo completo. Por lo general, se realiza una resonancia magnética local una vez al año. - A partir del año 6 en adelante, lo siguiente suele tener lugar una vez al año:
una radiografía con examen de laboratorio y una tomografía computarizada del tórax, así como una gammagrafía esquelética de todo el cuerpo y una resonancia magnética local.
Resumen
La enfermedad (sarcoma de Ewing) recibió su nombre de la primera descripción de James Ewing en 1921. Estos son tumores altamente malignos que surgen de células neuroectodérmicas primitivas degeneradas (= células precursoras inmaduras de células nerviosas). Por tanto, los sarcomas de Ewing pertenecen a los tumores sólidos primitivos, malignos.
Como se mencionó anteriormente, los sarcomas de Ewing afectan principalmente las áreas medias de los huesos tubulares largos y la pelvis, pero también es concebible una afección en la parte superior del brazo (= húmero) o en las costillas, por lo que aparecen paralelos al osteosarcoma. Debido a los signos de inflamación que lo acompañan, es posible la confusión con la osteomielitis.
Debido a las metástasis que ocurren muy rápidamente (aproximadamente ¼ de todos los pacientes ya muestran los llamados asentamientos secundarios en el momento del diagnóstico), los sarcomas de Ewing también se pueden encontrar en tejidos blandos, similares a los rabdomiosarcomas. Los pulmones suelen ser los más afectados por la metástasis.
Aún se desconocen las causas que podrían ser responsables del desarrollo del sarcoma de Ewing. Sin embargo, actualmente se asume que ni el componente genético (herencia) ni la radioterapia que ya se ha realizado pueden ser responsables del desarrollo. Sin embargo, se encontró que los sarcomas de Ewing a menudo ocurren cuando hay anomalías esqueléticas familiares o los pacientes sufren de retinoblastoma (= tumor de retina maligno que ocurre en la adolescencia) desde el nacimiento. La investigación ha demostrado que las células tumorales de la llamada familia de los sarcomas de Ewing muestran un cambio en el cromosoma número 22. Se supone que esta mutación (cambio genético) está presente en alrededor del 95% de todos los pacientes.
Los sarcomas de Ewing pueden causar hinchazón y dolor en la (s) región (es) afectada (s), que también pueden asociarse con alteraciones funcionales. También se pueden concebir fiebre y leucocitosis moderada (= aumento del número de leucocitos en la sangre). Debido a la posibilidad de confusión con la osteomielita, por ejemplo (ver arriba), un diagnóstico no siempre es fácil y, además de los procedimientos de imagen (examen de rayos X), puede ser necesaria una biopsia (= examen de tejido de una muestra de tejido).
El enfoque terapéutico aquí suele ser de varios niveles. Por un lado, el llamado plan de terapia generalmente prevé un tratamiento quimioterapéutico preoperatorio (= quimioterapia neoadyuvante). Incluso después de la extirpación quirúrgica del sarcoma de Ewing, se proporciona tratamiento de seguimiento terapéutico mediante radioterapia y, si es necesario, quimioterapia renovada. Aquí se nota una diferencia con el osteosarcoma: en comparación con el sarcoma de Ewing, el osteosarcoma tiene una menor sensibilidad a la radiación.
El hecho de que ocurran o no recurrencias (crecimiento tumoral renovado) depende en gran medida de la extensión de la metástasis, la respuesta a la quimioterapia preoperatoria y la "naturaleza radical" de la extirpación del tumor. Actualmente se cree que la tasa de supervivencia a cinco años es de alrededor del 50%. Las mejoras operativas en particular han permitido mejorar la probabilidad de supervivencia durante los últimos 25 años.








.jpg)




.jpg)