Síndrome de Ehlers-Danlos
Sinónimos

EDS, síndrome de Ehlers-Danlos-Meekeren, síndrome de Van Meekeren, fibrodisplasia elastica generalisata, dermatolisis, cutis hiperelastica, "piel de goma", etc.
Francés: Laxité articulaire congénitale multiple
Engl .: síndrome de Danlos, síndrome de Meekeren-Ehlers-Danlos, síndrome de Chernogubov, síndrome de Sack, síndrome de Sack-Barabas, síndrome de Van Meekeren I.
Ruso: síndrome de Chernogubov
Definición / introducción
los Síndrome de Ehlers-Danlos (EDS) resume un grupo de heterogéneos, genéticos Enfermedades del tejido conectivo juntos, causados por alteraciones en la síntesis de colágeno, una proteína estructural de la Tejido conectivo, síntomas condicionales y característicos de la piel, Articulaciones y órganos internos exposición.
frecuencia
los Síndrome de Ehlers-Danlos es raro. La prevalencia en la población total es 1: 5000; 90% de ellos están entre ellos Tipos I, II y III afectados (30% cada uno) y alrededor del 10% de tipo IV Las otras formas se observan raramente.
los Tipos I-III volverse dominancia autosómica heredado, es decir simplemente tiene que haber un gen defectuoso para que estalle la enfermedad. Los otros chicos lo harán autosómica recesivaes decir debe haber dos genes defectuosos, o Ligado al cromosoma Xes decir Transmisión de cromosomas sexuales, hereditaria.
historia
esto fue descrito por primera vez Síndrome de Ehlers-Danlos en el año 1668 de Job Janszoon de Meekeren (1611-1666), cirujano de Amsterdam. Había descubierto el síntoma de una sobreestirabilidad anormal en un español que podía llevar la piel de la barbilla hasta los ojos y sobre el pecho. Sin embargo, no observó ninguna otra anomalía.
primero 1891 creó el dermatólogo Chernogubov una descripción completa del cuadro clínico, incluida la afectación articular y vascular, por lo que en la medicina rusa
Literatura técnica hasta el día de hoy el nombre "Síndrome de Chernogubov" Es común.
Se siguieron más descripciones 1901 por el dermatólogo danés Eduard Ehlers (1863-1937) y 1908 por el dermatólogo de París Henri A. Danlos (1844-1912). No fue hasta 1933 que "Síndrome de Ehlers-Danlos" como prevalece el nombre de la enfermedad.
1949 hubo primeros conocimientos sobre la frecuencia familiar de la enfermedad y 1972 fue un error genético relacionado con eso Síndrome de Ehlers-Danlos descubierto. En 1986 se estableció una clasificación preliminar en 10 tipos, que se cambió en 1997 a una versión simplificada con la subdivisión en seis tipos principales.
causas
La causa de la enfermedad es un Defecto genético. Hay un cambio (mutación) en los genes que componen la proteína estructural. Colágeno describir en el genoma humano, el ADN. La mutación conduce a una estructura alterada y / o una síntesis reducida del colágeno, lo que conduce a una fuerza reducida de todo el tejido conectivo.
Ambos Tipos I y II es una mutación en el gen colágeno V, en el que Tipos IV una mutación en Colágeno III.
Síntomas
Debido a la síntesis de colágeno alterada y reducida, el Síndrome de Ehlers-Danlos las partes del cuerpo que son particularmente ricas en tejido conectivo: el piel, Articulaciones y Vasos sanguineos. Dado que el tejido conectivo carece de fuerza, se puede estirar demasiado y se desgarra muy rápidamente, lo que es especialmente cierto para los vasos sanguíneos que son demasiado pequeños, pero a veces demasiado. sangrado masivo puede conducir. Una complicación importante es la formación de protuberancias en los vasos, así llamados. Aneurismas con riesgo de rotura.
los Síntoma principal de la piel es el mas pronunciado Cutis hiperelásticaen el lado de la cuello, encima Articulaciones y tambien en cara se puede levantar hasta 4 cm o más. Después de soltarlo, inmediatamente vuelve a su posición inicial, razón por la cual recibió el nombre "Piel de goma" vistiendo. En general, la piel es notablemente fina (como el papel de fumar), suave y aterciopelada ("Piel de malvavisco").
Las heridas muestran un retraso en la cicatrización, por lo que las suturas tardan de 3 a 4 veces más en cicatrizar. Atróficos o hipertróficos, los inferiores a menudo se desarrollan a partir de las costuras. cicatriz. Además, existe la formación de protuberancias cutáneas llenas de líquido (suculentas) (pseudotumores mollusoides) en áreas muy estresadas del cuerpo, como Articulación de rodilla y codo, a la formación de tobilleras ("Almohadillas para los nudillos") en Dorso de la mano y del pie y de nódulos en el tacón.
los Las articulaciones se pueden estirar demasiado (Hiperflexibilidad), que a menudo se mueven en direcciones no deseadas y carecen de fuerza debido a los ligamentos articulares aflojados (Laxitud del ligamento). Esto puede provocar movimientos inusuales, como los que cabría esperar de "Contorsionistas" sabe. Las articulaciones tienden a Contorsiones (Dislocaciones) y malas posiciones. Aquellos se ven particularmente afectados hombro- y Articulaciones del tobillo, el Rótula (rótula), el Articulación temporomandibular (Articulación temporomandibular) y más raramente que Articulación del codo. La documentación de la sobremovilidad de las articulaciones (hipermovilidad) la realiza el Puntuación de Beightonque confirma hipermovilidad en 5 de los 9 puntos posibles.
Otros síntomas de las articulaciones son problemas articulares generalizados, dolor de cuello crónico, moverse- y Dolor de cadera, Conjunta y Dolores muscularesque son difíciles de tratar. A veces, los puntos de dolor ("Ponits tiernos"), que se definen como un área que reacciona dolorosamente a cargas de presión de 4 kg o menos. Además, existe un mayor riesgo de fracturas debido a la reducción de la masa ósea combinada con una estructura ósea anormal.
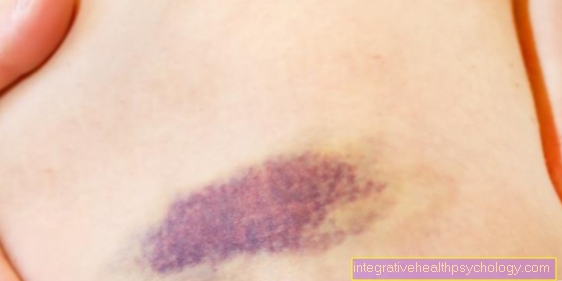
Debido a la fragilidad del tejido conectivo de los vasos sanguíneos, existe una tendencia pronunciada a Hematomas, espontáneamente o como resultado de un trauma,
principalmente en áreas con riesgo de lesiones. A esto le sigue un típico en las áreas afectadas. pigmentación marrón.
Después de las lesiones se observa una tendencia a sangrado prolongado con valores de coagulación normales. La fragilidad de los vasos sanguíneos más grandes puede ser provocada por el esfuerzo, accidentes, el embarazo o nacimiento provocar una hemorragia masiva que pone en peligro la vida.
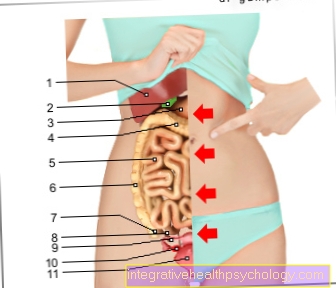
Dado que otras estructuras de tejido conectivo también son inferiores, puede volverse demasiado Vísceras (Hernia / hernia inguinal)), curvatura de la columna (Escoliosis), Grietas (rotura) del Intestinos y el útero (Útero), sacos (Aneurisma) de los vasos sanguíneos y la constricción de los pulmones por el aire libre en el pecho (Neumotórax) ven.
En casos raros, los cambios oculares están asociados con EDS como Astigmatismo (astigmatismo) o estrella verde (glaucoma) para observar.
diagnóstico
El diagnóstico se basa en el aspecto clínico, los síntomas y se complementa con un examen familiar (antecedentes familiares). Además Biopsia de piel en el que se examina el tejido cutáneo extraído con un microscopio electrónico y se evalúa su estructura de colágeno. La diferenciación en los diferentes tipos de Síndrome de Ehlers-Danlos se lleva a cabo mediante análisis de secuencia del ADN.
Clasificación / tipos
Tipo I, II: Chico clasico; Herencia: dominante autosómico; Principales síntomas: Hiperelasticidad y fragilidad de la piel, cicatrices atróficas, hipermovilidad articular; Causa: trastorno de la formación de colágeno V
Tipo III: Tipo hipermóvil; Herencia: dominante autosómico; Principales síntomas: hipermovilidad articular generalizada, afectación cutánea (hiperelasticidad y / o piel blanda y vulnerable); Causa: trastorno de la formación de colágeno V
Lea más sobre este tipo importante en: Síndrome de Ehlers-Danlos tipo III
Tipo IV: Tipo vascular; Herencia: dominante autosómico; Principales síntomas: piel fina translúcida, roturas de arterias, intestinos y útero, tendencia pronunciada al hematoma; Causa: trastorno de la formación de colágeno III
Tipo V: corresponde al tipo I.
Tipo VI: Tipo cifoescoliótico; Herencia: autosómica recesiva; Principales síntomas: disminución de la tensión del Musculatura ya al nacer"Bebé flojo"), retraso en el desarrollo de los reflejos de sujeción y apoyo, flexión lateral del Columna vertebral (Escoliosis), Causa principal: Deficiencia de hidroxilasa de Lsysl
Tipo VII A / B: Tipo artrocalastico; Herencia: dominante autosómico; Principales síntomas: hipermovilidad generalizada severa de las articulaciones con luxaciones repetidas, luxación congénita bilateral de cadera; Causa principal: Trastorno del colágeno tipo I
Tipo VII C: Tipo dermatosparactico; Herencia: dominante autosómico; Principales síntomas: fragilidad pronunciada de la piel, piel flácida, Causa principal: Deficiencia de peptidasa procolágeno I N-terminal
Terapia y profilaxis
Ninguno de los dos causal Aún un terapia sintomática Actualmente es posible, por lo que la profilaxis del daño consecuente está en primer plano. Deben evitarse lesiones y un mayor estrés en las articulaciones. Entonces debería estar seguro Deportesque están asociados con un mayor riesgo de lesiones no se ejercen. Debido al mayor riesgo de complicaciones el embarazo y nacimiento ambos Tipos I, II, IV y VI Se requiere una estrecha vigilancia.
También debe usarse con resfriados terapia supresora de la tos y en general a la Regulación de la consistencia de las heces. ser respetado, porque tal cosa Ruptura de colon (Rotura de colon) y un Neumotórax se puede evitar. A través de la fisioterapia temprana, especialmente en niños, las articulaciones que se pueden estirar demasiado se pueden estabilizar, lo que conduce al alivio de los síntomas de todo el sistema musculoesquelético.
Heridas Se debe tener especial cuidado y las operaciones solo deben realizarse en emergencias porque la cicatrización de heridas tarda de 3 a 4 veces más lenta de lo habitual.
pronóstico
Paciente con Síndrome de Ehlers-Danlos Suelen tener una esperanza de vida normal. Sin embargo, la enfermedad es progresiva, por lo que conduce a un deterioro cada vez mayor de la salud. Las heridas en la piel y las dislocaciones articulares afectan la calidad de vida del paciente, mientras que la rotura de los grandes vasos puede poner en peligro la vida.
Esperanza de vida
El síndrome de Ehlers-Danlos es una enfermedad crónica para la que todavía no existe un tratamiento causal y, por tanto, no tiene cura. Esto significa que, dado el estado actual de la tecnología médica, no hay forma de hacer nada sobre las causas del síndrome de Ehlers-Danlos y curarlo por completo. Lamentablemente, todavía no se pueden combatir y tratar los síntomas que se presentan. Solo se puede alentar al paciente en cuestión a que preste siempre atención en la vida cotidiana para no ejercer demasiada presión sobre las articulaciones y para evitar lesiones en la piel si es posible. Las intervenciones quirúrgicas solo deben realizarse en caso de emergencia y si no existen alternativas adecuadas.
En la mayoría de los casos es progresivo con un empeoramiento creciente de los síntomas y deficiencias en la vida diaria del paciente. Según el tipo de enfermedad, la enfermedad tiene diferentes efectos en la vida de los afectados. Los cambios en las articulaciones a veces conducen a la osteoartritis y la artritis en la primera infancia, por lo que los niños luego aprenden a caminar y sus pies pueden deformarse. Con el mayor riesgo de desprendimiento de retina o hemorragia de retina, la vista también se ve comprometida.
La intensidad con la que los síntomas se pronuncian en última instancia en cada individuo depende en gran medida del tipo de síndrome de Ehlers-Danlos y también pueden variar mucho dentro de los subtipos individuales. Para la mayoría de los tipos de síndrome de Ehlers-Danlos, la esperanza de vida es normal. En pacientes con síndrome de Ehlers-Danlos tipo IV, que afecta a los vasos, la esperanza de vida se reduce significativamente debido a complicaciones graves, como el riesgo de rotura espontánea de una arteria, especialmente la arteria principal (desgarro aórtico mediano) o el colon.
Es alrededor de 37 años para las mujeres y 34 años para los hombres.
También se puede suponer una esperanza de vida reducida con el síndrome de Ehlers-Danlos tipo VI.
.jpg)