Enfermedades por almacenamiento
definición
El término enfermedad de almacenamiento abarca una serie de enfermedades en las que determinadas sustancias se depositan en los órganos o células debido a procesos alterados en el metabolismo.
Dependiendo de la sustancia y el órgano, las enfermedades por almacenamiento pueden variar mucho en su gravedad y forma.
Algunas enfermedades por almacenamiento ya son evidentes al nacer y requieren tratamiento inmediato, mientras que otras solo aparecen a lo largo de la vida.

¿Qué enfermedades por almacenamiento existen?
-
Enfermedad por almacenamiento de hierro: hemocromatosis
-
Enfermedad por almacenamiento de cobre - Enfermedad de Wilson
-
Enfermedad por almacenamiento de proteínas
-
Enfermedad por almacenamiento de glucógeno
-
Enfermedad por almacenamiento lisosómico
-
Enfermedad por almacenamiento de ésteres de colesterol
-
Enfermedad por almacenamiento miocárdico
-
Enfermedad neutra por almacenamiento de grasa
Enfermedad por almacenamiento de hierro
La enfermedad por almacenamiento de hierro, conocida en los círculos de especialistas como hemocromatosis, es un trastorno metabólico en el que hay una mayor deposición de hierro en el cuerpo y en ciertos órganos.
En la mayoría de los casos, la enfermedad por almacenamiento de hierro es un defecto hereditario que conduce a una absorción excesiva de hierro del tracto gastrointestinal.
El exceso de hierro que se absorbe no puede excretarse tan rápidamente como se absorbe y, por lo tanto, se deposita en varios órganos.
Dependiendo del órgano afectado y la cantidad de hierro, pueden ocurrir una variedad de síntomas de hemocromatosis y molestias.
En casos raros, la enfermedad por almacenamiento de hierro también puede ocurrir como resultado de otra enfermedad subyacente o como resultado de transfusiones de sangre frecuentes, conocida como hemocromatosis secundaria.
El hierro adicional almacenado en los órganos, que normalmente no sirven como depósitos de hierro, conduce a procesos de remodelación.
Durante estos procesos de remodelación, se crea una forma de tejido cicatricial, que reemplaza el tejido del órgano sano y, por lo tanto, reduce la funcionalidad del órgano.
Con frecuencia, esto afecta principalmente a los órganos productores de hormonas en el abdomen, como el hígado (con mayor frecuencia) o el páncreas.
Órganos como el corazón, la piel y la glándula pituitaria también son algunos de los órganos dañados con mayor frecuencia.
El curso de la enfermedad suele ser insidioso y, por lo tanto, a menudo solo se nota en una etapa avanzada de la enfermedad.
Los síntomas dependen de la extensión del daño y de los órganos afectados.
Los síntomas generales como fatiga y cansancio son típicos al principio.
En el curso de la enfermedad, suele haber dolor articular en las articulaciones de los dedos, especialmente los dedos índice y medio, así como una coloración marrón notable de la piel.
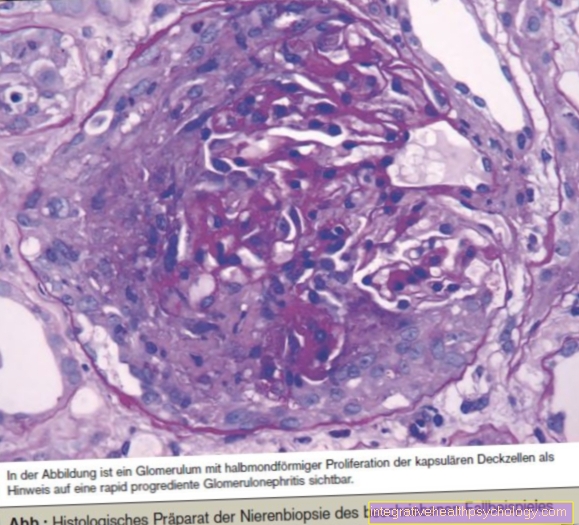
Con la ayuda de análisis de sangre y biopsias especiales de órganos individuales, se puede hacer un diagnóstico preciso de qué órganos están afectados y en qué medida.
Las manifestaciones orgánicas más comunes y típicas son principalmente el hígado con cirrosis hepática, que es un factor de alto riesgo para el desarrollo de cáncer de hígado, y el páncreas con el desarrollo de diabetes mellitus.
Las opciones para tratar la enfermedad por almacenamiento de hierro se limitan a la excreción regular del exceso de hierro.
Aún no se conoce una cura causal.
En primer lugar, se recomienda una dieta baja en hierro, así como el consumo regular de té negro, ya que esto reduce la absorción de hierro en los intestinos.
Si los valores de hierro están elevados a pesar de una dieta baja en hierro, la sangría es el método de elección.
Aquí, se extraen 500 ml de sangre del paciente, por lo que se pierde el hierro unido a las células sanguíneas.
Después de alcanzar el nivel objetivo de hierro en la sangre, se recomienda realizar una sangría cada 2-3 meses con controles de laboratorio minuciosos.
Esta intervención a menudo se puede prescindir en mujeres en edad fértil, ya que el sangrado menstrual conduce a una pérdida suficiente de hierro.
Como alternativa a la sangría, también se encuentran disponibles medicamentos que se unen al hierro, pero estos solo se usan si la sangría no es factible, por ejemplo, debido a anemia, anemia u otra enfermedad subyacente.
Con un diagnóstico temprano y una terapia constante, los afectados por la enfermedad por almacenamiento de hierro tienen una esperanza de vida normal.
Enfermedad por almacenamiento de cobre
La enfermedad por almacenamiento de cobre, la llamada enfermedad de Wilson, es una enfermedad metabólica que se basa en la excreción alterada de cobre.
La razón de esto es un defecto genético hereditario en una proteína que prepara el cobre para su excreción en la bilis.
Si hay un defecto aquí, el cobre ya no se puede excretar en cantidades suficientes.
Se acumula en el torrente sanguíneo y, como resultado, se deposita en varios órganos.
Normalmente, el cobre se deposita principalmente en el hígado, la córnea, los glóbulos rojos y el cerebro.
En particular, la participación del hígado y el cerebro, en combinación, conduce a síntomas típicos, que conducen al diagnóstico de sospecha de una enfermedad por almacenamiento de cobre.
Los primeros síntomas suelen aparecer entre los 5 y los 10 años, por ejemplo en forma de inflamación del hígado, conocida como hepatitis, o restricciones neurológicas debidas a una función hepática disminuida, como somnolencia y manos temblorosas.
A partir de los 10 años, suelen aparecer molestias neurológicas, como temblores finos en las manos, demencia, trastornos de la deglución o del habla y trastornos de la marcha.
Además, los depósitos de cobre pueden volverse visibles en el ojo.
Aquí hay un anillo verde-marrón en la córnea.
El diagnóstico de la enfermedad por almacenamiento de cobre se puede confirmar con la ayuda de análisis de sangre y orina, posiblemente una biopsia del hígado y varias pruebas de imagen.
Si se confirma el diagnóstico de enfermedad por almacenamiento de cobre, la terapia primaria consiste en una combinación de una dieta baja en cobre y medicamentos que sirven para excretar cobre, los llamados agentes quelantes, por ejemplo, D-penicilamina.
Si la terapia se inicia temprano y de manera constante, el pronóstico de la enfermedad es bueno.
Lo único importante es hacer un diagnóstico temprano, antes de que se produzcan daños en los órganos debido a los depósitos de cobre.
Se aplica lo siguiente: toda enfermedad hepática poco clara que no se deba a una infección, en combinación con trastornos del movimiento poco claros antes de los 45 años, debe aclararse con respecto a una enfermedad por almacenamiento de cobre.
Lea también el artículo: La prueba genética
Enfermedad por almacenamiento de proteínas
La denominada enfermedad por almacenamiento de proteínas no es un cuadro clínico reconocido según la Organización Mundial de la Salud.
Más bien, es un concepto que el Prof.Dr. Lothar Wendt fue desarrollado y publicado.
En su trabajo, el profesor Wendt siguió un enfoque alternativo para explicar las enfermedades comunes en nuestra sociedad, contrastando la visión de la medicina tradicional con la pregunta del "por qué".
Un ejemplo típico de este enfoque puede ilustrarse con la diabetes, una enfermedad común.
En la diabetes mellitus tipo 2, el nivel de azúcar en sangre es muy alto.
Estos niveles elevados de azúcar en sangre provocan daños en todo el cuerpo con graves complicaciones.
Por lo tanto, el enfoque médico convencional consiste en reducir constantemente el nivel de azúcar en sangre para evitar daños mayores.
El profesor Wendt, por otro lado, pregunta en su concepto de trabajo por qué ocurren estos niveles elevados de azúcar en sangre y si la razón de esto podría ser una compensación.
Aquí presenta la teoría de que los depósitos de proteínas en las paredes de los vasos sanguíneos hacen que se espesen.
El profesor Wendt explica que el aumento del nivel de azúcar en sangre es una reacción al engrosamiento de las paredes de los vasos sanguíneos para transportar una cantidad suficiente de azúcar a la célula a pesar del aumento de la resistencia y la vía de difusión más larga.
Según Wendt, no es el azúcar el factor que causa la enfermedad, sino la proteína y, en última instancia, el término diabetes es engañoso.
El término aumento del nivel de azúcar en sangre como resultado de una enfermedad causal de almacenamiento de proteínas sería más apropiado según su concepto.
En la actualidad, sin embargo, hay una falta de estudios basados en evidencia que apoyen este enfoque explicativo y el concepto de enfermedad.
Solo en el tratamiento de la osteoartritis ya hay afectados en algunos grupos de autoayuda que informan que han aliviado o incluso eliminado la osteoartritis mediante la terapia de degradación de proteínas dirigida.
Sin embargo, debe tenerse en cuenta que se trata de valores empíricos individuales sin un grupo de referencia, que solo tuvieron éxito si la terapia se inició en una etapa muy temprana de la enfermedad.
Profesores destacados de varios departamentos no ven, teniendo en cuenta la situación actual del estudio, ninguna evidencia de la exactitud del concepto de enfermedad por almacenamiento de proteínas del Prof. Wendt.
Enfermedad por almacenamiento de glucógeno
En las enfermedades por almacenamiento de glucógeno, un defecto genético hereditario conduce a una deposición excesiva de glucógeno en el cuerpo.
El glucógeno también se conoce coloquialmente como almidón de hígado.
Se trata de una molécula de glucosa larga y ramificada, que se almacena en el hígado en particular y sirve como proveedor del azúcar portador de energía.
Hay un total de nueve formas diferentes de enfermedad por almacenamiento de glucógeno, cada una de las cuales se basa en un defecto genético diferente y conduce al depósito de glucógeno en diferentes órganos.
Las formas más comunes incluyen la enfermedad por almacenamiento de glucógeno tipo I, la enfermedad de von Gierke, la enfermedad por almacenamiento de glucógeno tipo II, la enfermedad de Pompe y la enfermedad por almacenamiento de glucógeno tipo V, enfermedad de McArdle.
Las diversas formas difieren tanto en sus síntomas como en el inicio de la enfermedad.
La enfermedad por almacenamiento de glucógeno tipo I suele notarse por un hígado agrandado y un abdomen distendido, además de que a menudo hay convulsiones y tendencia a sangrar.
En la enfermedad por almacenamiento de glucógeno tipo II, la atrofia muscular en todo el cuerpo y una lengua demasiado grande son particularmente notables.
En la enfermedad por almacenamiento de glucógeno tipo V, también se produce un desgaste muscular generalizado, pero en combinación con dolor muscular y calambres después de un esfuerzo.
La terapia de las enfermedades por almacenamiento de glucógeno depende del tipo de enfermedad y su gravedad.
Enfermedad por almacenamiento lisosómico
El término enfermedad de almacenamiento lisosómico abarca un gran grupo de enfermedades que se basan en un defecto genético en los lisosomas.
Los lisosomas son un grupo de células del cuerpo humano que actúan como el estómago o el cubo de basura de las células.
Todos los componentes celulares en exceso y los productos de desecho de la célula se descomponen en los lisosomas.
Si los lisosomas son defectuosos, estos productos de desecho celular se acumulan, que luego se depositan tanto en la célula como en otros órganos.
45 enfermedades están actualmente asignadas al grupo de enfermedades de almacenamiento lisosómico.
La mayoría de las enfermedades son variantes muy raras de la enfermedad por almacenamiento.
Las formas más comunes de enfermedad por almacenamiento lisosómico son la enfermedad de Gaucher y la enfermedad de Fabry.
Leer más sobre el tema: Enfermedad de Fabry
En la enfermedad de Gaucher, los procesos de degradación interrumpidos conducen a una acumulación de grasas en las células y otros órganos.
Los síntomas varían ampliamente debido a la posibilidad de afectar a todo el cuerpo.
Son típicos el agrandamiento del hígado y el bazo, trastornos en el sistema de formación de sangre y convulsiones.
La enfermedad a menudo se nota en la infancia debido a un trastorno de la alimentación.
La enfermedad de Fabry, por otro lado, es significativamente más rara que la enfermedad de Gaucher y afecta principalmente a los niños debido a su herencia.
Los síntomas de la enfermedad de Fabry inicialmente incluyen ataques de ardor de dolor en los dedos, molestias gastrointestinales y opacidad corneal.
Más tarde, el corazón puede infectarse con insuficiencia cardíaca y accidentes cerebrovasculares.
Obtenga más información sobre el tema aquí: Enfermedad de Gaucher.
Enfermedad por almacenamiento de ésteres de colesterol
La enfermedad por almacenamiento de ésteres de colesterol pertenece al grupo de enfermedades por almacenamiento lisosómico, que es una enfermedad metabólica hereditaria rara.
La enfermedad de los ésteres de colesterol se basa en un defecto en la lipasa ácida lisosomal, que normalmente descompone las grasas como los ésteres de colesterol y los triacilglicéridos.
La descomposición reducida de estas grasas conduce a una acumulación de grasas en la célula y, en consecuencia, también en la circulación del cuerpo.
Esta enfermedad no causa molestias durante mucho tiempo, solo el agrandamiento reactivo del hígado puede provocar una sensación de presión en la parte superior derecha del abdomen, náuseas o hinchazón.
Los análisis de sangre muestran valores sanguíneos elevados de colesterol y lípidos, así como valores reducidos de grasas buenas (HDL).
Puede aparecer hígado graso en la ecografía de la parte superior del abdomen.
El tratamiento de la enfermedad por almacenamiento de ésteres de colesterol se lleva a cabo con fármacos que inhiben la captación de colesterol con colestiramina o ezetimiba y, además, reducen los valores de lípidos en sangre con estatinas como simvastatina.
Enfermedad por almacenamiento miocárdico
En la enfermedad por almacenamiento miocárdico, los productos de degradación se depositan en las paredes del corazón, lo que puede restringir gravemente el rendimiento y la función de bombeo del corazón.
Dos enfermedades de almacenamiento diferentes pueden conducir a estos depósitos en las paredes del corazón: la enfermedad de Fabry por almacenamiento lisosómico, rara y hereditaria, y la llamada amiloidosis.
En la enfermedad de Fabry, un defecto genético heredado conduce a una descomposición reducida de los productos metabólicos, que como resultado se depositan, entre otras cosas, en las paredes del corazón y pueden provocar daños graves.
La amiloidosis, por otro lado, puede ser tanto hereditaria como adquirida a lo largo de la vida.
Con este cuadro clínico, también, hay depósitos de productos metabólicos alterados anormalmente que, además de otros órganos, se acumulan principalmente en el corazón y aquí limitan severamente la función del corazón.
Una enfermedad de almacenamiento miocárdico se hace evidente al principio por síntomas generales como debilidad y fatiga.
Con el tiempo, hay una falta de aire cada vez mayor después del ejercicio y, en algún momento, también en reposo.
El agua en los pulmones, el abdomen, las piernas y el pericardio son efectos secundarios típicos a medida que avanza la enfermedad.
Los procedimientos de imágenes y una biopsia del músculo cardíaco son necesarios para un diagnóstico claro de la enfermedad por almacenamiento miocárdico.
La terapia posterior se basa entonces en la enfermedad subyacente que la causó.
Enfermedad neutra por almacenamiento de grasa
Las enfermedades neutrales por almacenamiento de grasas son enfermedades muy raras en las que la degradación y el almacenamiento de una grasa, los llamados triglicéridos, son defectuosos.
Hasta la fecha, solo se han descrito 50 casos de enfermedades neutrales por almacenamiento de grasa en todo el mundo.
Como ocurre con la mayoría de las enfermedades por almacenamiento, la causa del defecto genético también es hereditaria en la enfermedad por almacenamiento de grasa neutra.
La enfermedad a menudo se nota en la primera infancia debido a un trastorno del desarrollo.
La mayoría de los afectados desarrollan un hígado agrandado con una disfunción hepática asociada, así como problemas oculares y pérdida auditiva.
La atrofia muscular y los trastornos de la marcha pueden ocurrir en edades avanzadas.